Regulatory Affairs Consultancy

Regulatory Affairs Consultancy
Partnering with Talouf helps you save valuable time and effort while accelerating your time to market. We are committed to ensuring your smooth adherence to the Saudi Food and Drug Authority (SFDA) regulations. Our team assists you in staying updated with the latest standards to maintain your product compliance.
Medical Device
Product Classification Application on PCS:
- This application is essential for manufacturers seeking to get their Medical Devices products into Saudi Arabia, as it aids in determining the appropriate classification for their devices, clearing the necessary steps required for registering these products in strict accordance with regulations and requirements of Saudi Food and Drug Authority SFDA
The Manufacturer is responsible for classifying the product according to SFDA rules and creating the necessary technical files.


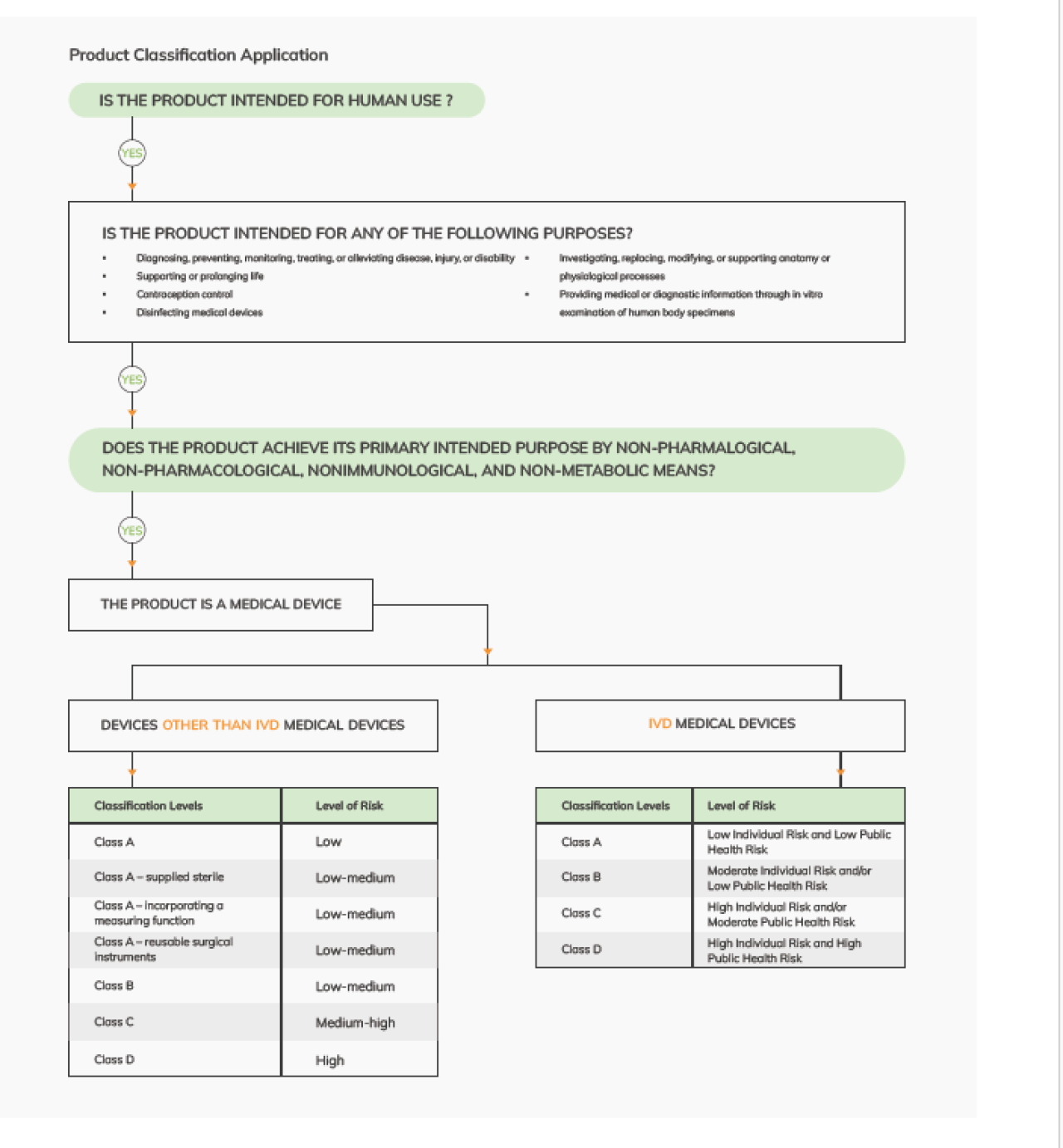
Classification of Devices other than IVD Medical Devices
- Manufacturer ascertaining the device's classification by employing a set of classification rules grounded in:
- Intended Use: this information is typically obtained from:
Instructions for Use (IFU), Label, Manufacturer's advertising
materials, and technical documentation - Risk Assessment
- Degree of invasiveness in the human body
- Duration of Use
Classification of IVD Medical Devices
- Manufacturer ascertaining the device's classification by employing a set of classification rules grounded in:
- Using the classification rules for IVD medical devices, and
- Considering both the: Intended use of the device, Level of risk to the patient and public of an incorrect result.
Authorized
Representation (AR)
- Authorized Representation (AR) refers to a Saudi-based entity that has entered into a contractual regulatory agreement with a medical device manufacturer. This agreement designates the entity to act as the manufacturer's representative within the Kingdom of Saudi Arabia.
- Any foreign manufacturers looking to enter the Saudi Arabian market are required to appoint a representative within Saudi Arabia who has the legal authority to act on their behalf in dealings with the SFDA. It helps them navigate complex regulatory requirements, ensures compliance with SFDA standards, and facilitates effective communication with the SFDA, ultimately enabling them to offer safe and compliant products to the Saudi market standards.

Why appoint Talouf as your Authorized
Representation (AR)?
Regulatory Compliance
Ensuring that your products or medical devices comply with the SFDA's regulations and standards.
Registration and Documentation
Assisting you in the registration process of your medical devices with the SFDA and maintain accurate records of all related documentation.
Labeling and Packaging
Ensuring that product labeling and packaging adhere to SFDA requirements, including language and safety information.
Communication with SFDA
As an Authorized Representative, we serve as the primary point of contact between the foreign manufacturer and the SFDA, and handle all matters related to registration requirements, including document review, ensuring compliance with regulations, addressing regulatory issues, and submitting documents to the Saudi Food and Drug Authority (SFDA)
Post-Market Surveillance
Post-market surveillance and reporting adverse events or product recalls to the SFDA, if applicable.
Quality Assurance
Involved in ensuring that products meet quality and safety standards and coordinate with the SFDA for inspections and audits.
Manufacturers are presented with two distinct options when appointing an Authorized Representative (AR)
Agreement between a single legal manufacturer and an authorized representative
Agreement between an organization representing multiple legal manufacturers within a single company and an authorized representative
Required Authentication Steps
Manufacturers Outside KSA
Hague Convention Members
“Apostille” that been issued by an accredited body in the foreign country.
Non-Hague Convention Countries
- Chamber of Commerce in foreign country.
- The Ministry of Foreign Affairs in foreign country.
- The Saudi embassy in the foreign country.
- The Saudi Foreign Ministry.
Manufacturers Within KSA
- Chambers of Commerce and Industry.
- The Embassy of the Foreign Party in the Kingdom.
- The Saudi Foreign Ministry.
Medical Device Marketing Authorization (MDMA)
In adherence to SFDA regulations, obtaining a Medical Device Market Authorization (MDMA) is a mandatory request for Manufacturers to market medical devices, including IVDs, in Saudi Arabia.
Requirements
To obtain Medical Device Marketing Authorization in compliance with SFDA regulations, manufacturers must adhere to the following key requirements:
Quality and Safety Compliance with SFDA regulations.
Submit Technical Documentation for Medical Device and/or IVD Medical Device
Foreign manufacturers must appoint an Authorized Representative within Saudi Arabia who will be responsible for regulatory matters through the GHAD System - Licensing Services
Local Manufactures: License of medical devices manufacture through the GHAD System - Licensing Services
Detailed Device Description and Specifications
Manufacturer's Obligatory Information
Checklist of Essential Principles
Proof of Compliance with Relevant Essential Principles
Risk Management File
Post-Market Surveillance
Licensing Requirements for Medical Devices
Medical Device Establishment License
(MDEL)
The Medical Device Establishment License (MDEL) is a regulatory requirement established by the Saudi Food and Drug Authority (SFDA) to ensure the safety, efficacy, and quality of medical devices distributed or manufactured in Saudi Arabia. It is a critical step in the regulatory process to monitor and control the medical device market within the country.
Requirements
Quality Management System (QMS): ISO 13485
Good Distribution Practices (GDP): For distributors, adherence to Good Distribution Practices is crucial. This includes proper storage, handling, and distribution of medical devices to maintain their integrity and safety.
Good Manufacturing Practices (GMP): For manufacturers, compliance with Good Manufacturing Practices is essential to ensure the production of safe and effective medical devices.
Technical Documentation.
Inspections and Audits: Be prepared for periodic inspections and audits by the SFDA to ensure ongoing compliance.
Medical Device Importation License
(MDIL)
The purpose of this document is to specify and clarify the requirements for the importation and shipment clearance of medical devices/supplies. This document applies to medical devices, supplies, and their associated accessories intended for importation for special use.
Requirements
The MDIL for specific purposes is typically sought when an importer has a specialized or unique requirement for importing specific medical devices or equipment that may not fall under standard licensing categories. It allows for the importation of medical devices for specific activities, such as research, Demonstration purposes, clinical trials, or other specialized healthcare initiatives.
The application to be submitted to the SFDA should include comprehensive details, including the specific purpose, the type and the quantity of medical devices intended for importation, and their intended use
Importers must demonstrate that the medical devices to be imported meet the necessary safety and quality standards, even for specialized purposes.
Unique Device Identification (UDI)
UDI system objects to document unique devices codes for medical devices based on accredited international standards, with the purpose of allowing all stakeholders to identify medical devices information through the unique device identification code that is registered on the system.
Manufacturers and their authorized representatives are directly responsible for submitting, maintaining, and modifying all data of unique device identification codes for their medical devices according to the SFDA guidance for UDI.
Advantages of Unique Device Identification (UDI)
Standardized Identification
Ensures consistent device identification for streamlined processes.
Device Traceability
Enhances monitoring and control of devices throughout their lifecycle.
Fraud Prevention
Facilitates identification of counterfeit devices, bolstering patient safety.
Data Management
Supports effective management of recalls, corrective actions, and adverse events.
Lifecycle Efficiency
Establishes controls for efficient device management from inception to disposal.
Achieving Safety
Promotes device safety for improved healthcare outcomes.

Dr. Rayan Harb
Business Consultancy
Dr. Rayan Harb
Hey, how can I help you today?
Powered by Elementor